
ในยุคที่การระบาดของโรคและการดื้อยากลายเป็นความท้าทายด้านสาธารณสุขระดับโลก การย่นเวลาการค้นพบและออกแบบยาต้านไวรัสกลายเป็นหัวใจสำคัญของการตอบโต้เชิงวิทยาศาสตร์ สถิติทั่วไปชี้ว่ากระบวนการพัฒนายาแบบดั้งเดิมอาจกินเวลา 10–15 ปีและมีต้นทุนหลายร้อยล้านถึงพันล้านดอลลาร์สหรัฐ ทำให้การนำ AI มาเร่งขั้นตอนสำคัญกลายเป็นทางเลือกที่น่าจับตามอง ในบริบทนี้ สตาร์ทอัพสิงคโปร์ DeNovoDrug เปิดตัวแพลตฟอร์มที่ผสานข้อดีของ diffusion model กับ Graph Neural Networks (Graph‑NN) เพื่อออกแบบโมเลกุลต้านไวรัสโดยอาศัยการสร้างตัวอย่างโมเลกุลเชิงนวัตกรรมและการประเมินเชิงโครงสร้างแบบกราฟ ผลลัพธ์เริ่มต้นชี้ให้เห็นว่าขั้นตอนออกแบบโมเลกุลและการคัดกรองเบื้องต้นสามารถย่นจากปีเหลือเป็นเดือนหรือแม้แต่สัปดาห์ในบางกรณี
บทความนี้จะพาไปรู้จักภาพรวมของเทคโนโลยีที่ DeNovoDrug นำมาใช้ อธิบายสถาปัตยกรรมของระบบ—รวมถึงการประสานงานระหว่าง diffusion-based generative models และ Graph‑NN สำหรับการคาดการณ์คุณสมบัติทางชีวภาพ—พร้อมนำเสนอเคสสตั๊ดดี้จากงานนำร่องและผลการทดสอบเบื้องต้น ตลอดจนสรุปแนวทางปฏิบัติสำหรับนักวิจัยที่ต้องการนำแนวทางนี้ไปปรับใช้จริง ทั้งข้อดี ข้อจำกัด และประเด็นด้านการตรวจสอบความถูกต้องทางชีวภาพและการกำกับดูแลที่ยังต้องให้ความสำคัญ
บทนำ: ทำไมการเร่งค้นพบยาถึงสำคัญ
บริบทปัญหาของกระบวนการค้นพบยาแบบดั้งเดิม
กระบวนการค้นพบยาแบบดั้งเดิมยังคงเป็นความท้าทายทั้งในแง่เวลา ต้นทุน และความเสี่ยงทางวิทยาศาสตร์ โดยทั่วไปแล้วการนำยาใหม่จากแนวคิดสู่การวางจำหน่ายใช้เวลาประมาณ 10–15 ปี และมีต้นทุนรวมในการวิจัยและพัฒนาอยู่ที่ประมาณ 1–2 พันล้านดอลลาร์สหรัฐฯ ต่อผลิตภัณฑ์หนึ่งชนิด ผลที่ตามมาคือทรัพยากรทางการเงินและเวลาที่มหาศาลต้องถูกจัดสรรให้กับโครงการที่หลายโครงการอาจไม่สำเร็จ
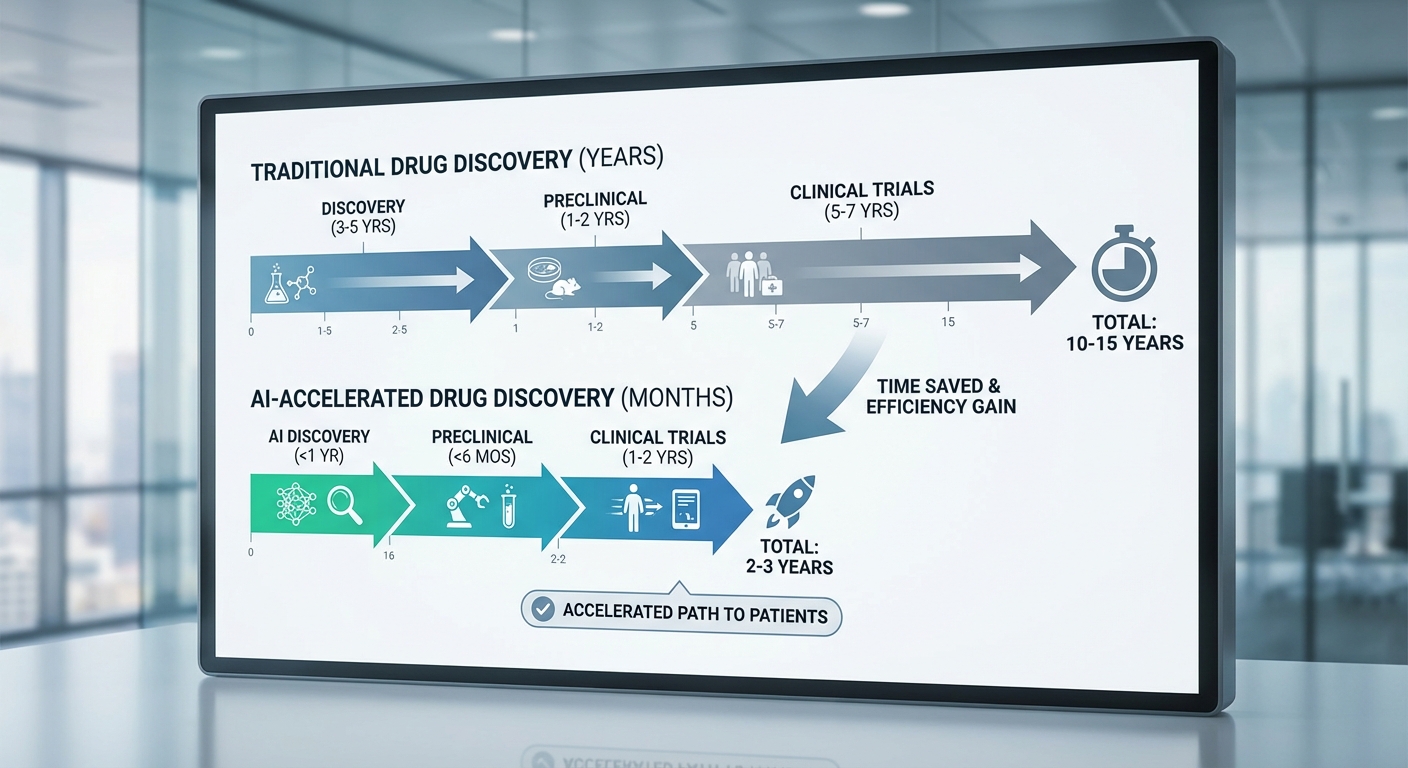
นอกจากเรื่องเวลาและต้นทุนแล้ว อัตราความล้มเหลวของ candidate ทางยาก็สูงมาก—มีการประมาณการว่า น้อยกว่า 1% ของโมเลกุลที่เริ่มต้นเข้าสู่กระบวนการค้นพบจะไปถึงการวางตลาดจริง นี่หมายความว่าบริษัทและนักลงทุนต้องรับความเสี่ยงแบบกระจัดกระจาย การทดสอบทางชีวภาพ การสังเคราะห์ และการทดลองในสัตว์และมนุษย์ล้วนใช้ทรัพยากรอย่างหนัก และการคัดกรองแบบเดิมมักช้าและมีประสิทธิภาพจำกัด
บทบาทของ DeNovoDrug และความหมายของการลดเวลาเป็น 'ไม่กี่เดือน'
ในบริบทนี้ แพลตฟอร์ม DeNovoDrug ของสตาร์ทอัพสิงคโปร์ตั้งเป้าหมายที่จะเปลี่ยนโครงสร้างพื้นฐานของการค้นพบยาโดยเฉพาะการออกแบบโมเลกุลต้านไวรัส ด้วยการผสานเทคโนโลยี Diffusion Model และ Graph‑Neural Networks (Graph‑NN) แพลตฟอร์มมุ่งลดระยะเวลาของขั้นตอนการค้นพบเริ่มต้น—เช่น การสร้างและคัดกรองโมเลกุลนำ (hit generation) และการปรับปรุงนำเป็นตัวนำหลัก (lead optimization)—จากปีหรือหลายปีให้เหลือเพียง ไม่กี่เดือน
การกล่าวว่าเทคโนโลยีสามารถลดเวลาเป็น "ไม่กี่เดือน" มิได้หมายถึงการขออนุญาตนำยาเข้าสู่ตลาดในช่วงเวลาเดียวกัน แต่มีความหมายสำคัญเชิงกลยุทธ์ดังนี้:
- เร่งวงจรการค้นพบให้สามารถสร้างและทดสอบชุดโมเลกุลเชิงออกแบบได้ภายในไตรมาสเดียว ทำให้สามารถทำ A/B testing ของโมเลกุลและสมมติฐานทางชีวภาพได้เร็วขึ้น
- ลดต้นทุนต่อชุดโมเลกุลที่ต้องทดสอบในห้องปฏิบัติการและการทดลองเบื้องต้น ซึ่งลดความเสี่ยงด้านการเงินและเพิ่มความยืดหยุ่นของพอร์ตโฟลิโอ
- เพิ่มความสามารถในการตอบสนองต่อการระบาดของโรคใหม่หรือการกลายพันธุ์ของไวรัสอย่างทันท่วงที เพราะสามารถสร้างและเสนอโมเลกุลต้านไวรัสเบื้องต้นได้ภายในเดือนแทนที่จะเป็นปี
สำหรับภาคธุรกิจและระบบสาธารณสุข ผลลัพธ์ที่ได้จะเป็นการเปลี่ยนแปลงเชิงโครงสร้าง: องค์กรสามารถย่นเวลาการตัดสินใจเชิงวิทยาศาสตร์ ลดความจำเป็นในการลงทุนล่วงหน้าจำนวนมาก และเร่งการส่งมอบนวัตกรรมไปสู่การทดสอบทางคลินิกได้เร็วขึ้น อย่างไรก็ตาม การนำโมเดลเชิงคอมพิวเตอร์มาใช้ต้องจับคู่กับการยืนยันเชิงทดลองและการประเมินตามกรอบกฎระเบียบเพื่อให้แน่ใจในความปลอดภัยและประสิทธิผลก่อนเข้าสู่ตลาดจริง
ภาพรวมเทคโนโลยี: Diffusion Models และ Graph Neural Networks
Diffusion Models — หลักการพื้นฐานและการสร้างตัวอย่าง
Diffusion models เป็นกลุ่มของโมเดลการสร้างตัวอย่าง (generative models) ที่เรียนรู้การย้อนสัญญาณรบกวน (denoising) ของข้อมูล โดยหลักการสำคัญแบ่งเป็นสองขั้นตอนคือการเพิ่มสัญญาณรบกวนแบบก้าวต่อก้าว (forward noising) และการเรียนรู้การย้อนกลับเพื่อนำสัญญาณรบกวนออก (backward denoising) เพื่อสร้างตัวอย่างใหม่จากเสียงรบกวนบริสุทธิ์ ขั้นตอนการฝึกมักจะประมาณฟังก์ชันคะแนน (score function) ซึ่งแทนค่าเชิงเวกเตอร์ของความชันของความหนาแน่นความน่าจะเป็น ∇ log p(x) เพื่อชี้นำการย้อนคืนสู่ตัวอย่างที่มีความเป็นจริง
ในทางปฏิบัติ กระบวนการ forward จะค่อย ๆ เติม noise เข้าไปในตัวอย่างจริงจนกลายเป็นสัญญาณสุ่ม ส่วนกระบวนการ backward จะใช้โมเดลที่ฝึกแล้วค่อย ๆ ลด noise ออกในทิศทางที่เพิ่มความเป็นไปได้ของข้อมูล โมเดลชุดนี้ถูกพิสูจน์ว่าสร้างตัวอย่างที่มีความหลากหลายและคุณภาพสูง และมัก มีประสิทธิภาพในการสร้างตัวอย่างแบบต่อเนื่อง (continuous data) เช่น รูปภาพ เสียง หรือตัวแทนเชิงตัวเลขของโครงสร้างโมเลกุล
ตัวอย่างเชิงปริมาณ: งานวิจัยล่าสุดแสดงให้เห็นว่า diffusion models สามารถทำได้ใกล้เคียงหรือเหนือกว่าผลงานของ GANs ในหลายโดเมน และเทคนิคการลดจำนวนขั้นตอนการสุ่ม (เช่น DDIM หรือการใช้ SDE แบบพัฒนาแล้ว) ช่วยลดเวลา sampling จากหลักพันขั้นตอนเหลือไม่กี่สิบขั้นตอนในบางการใช้งาน
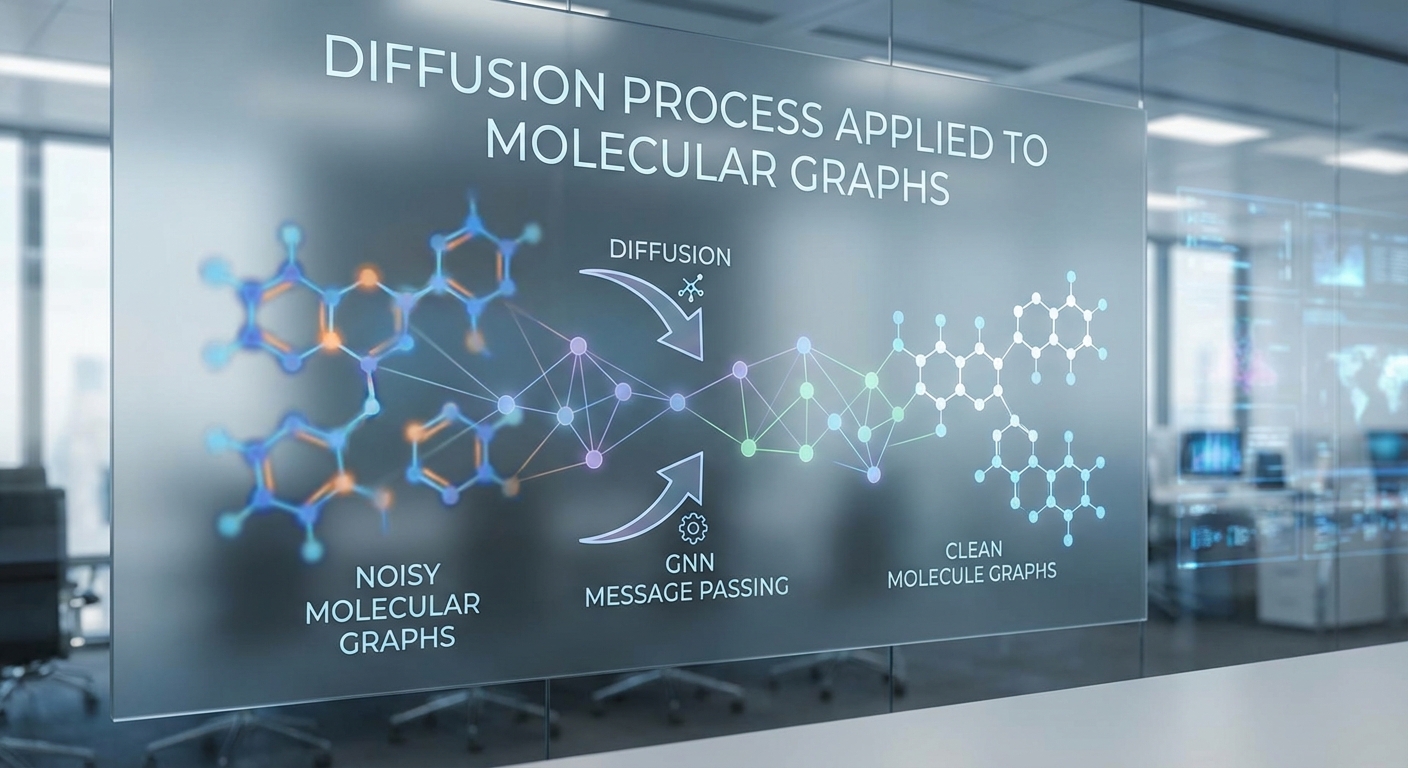
Graph Neural Networks (GNN) — ตัวแทนเชิงโครงสร้างของโมเลกุล
Graph Neural Networks (GNN) เป็นสถาปัตยกรรมที่ออกแบบมาเพื่อประมวลผลข้อมูลที่มีโครงสร้างแบบกราฟ ซึ่งโมเลกุลเป็นตัวอย่างที่เหมาะสมเนื่องจากประกอบด้วยโหนด (อะตอม) และขอบ/พันธะ (bonds) โดยหลักการของ GNN พื้นฐานคือการส่งผ่านข้อความ (message passing) ระหว่างโหนด:
- แต่ละโหนดมีเวกเตอร์สถานะ (node features) ที่แทนลักษณะของอะตอม เช่น ธาตุ สถานะอิเล็กตรอน ฯลฯ
- ขอบระหว่างโหนดมีคุณสมบัติ (edge features) เช่น ประเภทพันธะ ความยาวมุม ฯลฯ
- ในการอัปเดตแต่ละรอบ โหนดจะรวบรวมข้อความจากเพื่อนบ้าน (aggregation) แล้วอัปเดตสถานะของตนผ่านฟังก์ชันอัพเดต (update functions)
ผลลัพธ์คือการได้ตัวแทนเชิงฟีเจอร์ที่รักษาโครงสร้างเชิงสัมพันธ์ของโมเลกุลไว้ ทำให้ GNN เหมาะสำหรับงานทำนายคุณสมบัติทางเคมี (property prediction), การจำแนกปฏิกิริยา และการสร้างโมเลกุลใหม่แบบเชิงโครงสร้าง
การผสาน Diffusion Models กับ GNN — เหตุผลและข้อได้เปรียบเชิงปฏิบัติ
การรวมสองเทคนิคนี้เข้าด้วยกันทำให้ได้ระบบที่สามารถสร้างโมเลกุลใหม่ ๆ ได้ทั้งในเชิงความเป็นจริงของโครงสร้างและการควบคุมคุณสมบัติทางเคมี โดยแนวทางที่พบว่าได้ผลดีคือการใช้ diffusion model ใน latent/feature space ของกราฟ ซึ่งสร้างจาก GNN ดังนี้:
- การแทนโมเลกุลใน latent space: GNN ถูกใช้แปลงกราฟโมเลกุลเป็นตัวแทนเชิงฟีเจอร์ (embeddings) ที่จับความสัมพันธ์ของอะตอมและพันธะ การทำ diffusion บน latent space นี้ช่วยให้การ sampling เกิดขึ้นในพื้นที่ต่อเนื่องและมีโครงสร้าง ซึ่งง่ายต่อการเรียนรู้การเคลื่อนที่ของความน่าจะเป็น
- การควบคุมคุณสมบัติ (property conditioning): เนื่องจาก latent space แทนข้อมูลเชิงฟีเจอร์ของโมเลกุลได้ดี การใส่เงื่อนไข (เช่น ค่าการยึดติดกับโปรตีน เป้าหมายความเป็นพิษ สภาพละลายน้ำ) ทำได้โดยตรงผ่านการปรับคะแนนใน diffusion process ผลคือสามารถออกแบบโมเลกุลที่ตอบโจทย์คุณสมบัติเฉพาะได้มากขึ้น
- การบูรณาการโครงสร้างและความต่อเนื่อง: GNN ดูแลความถูกต้องเชิงโครงสร้างของโมเลกุล ส่วน diffusion model ดูแลการสร้างตัวอย่างที่มีความต่อเนื่องและหลากหลาย ทำให้ได้โมเลกุลที่ทั้งสมจริงและมีความเป็นไปได้ทางเคมีสูง
ตัวอย่างเชิงแนวทางปฏิบัติ: แนวทาง latent diffusion บนกราฟช่วยลดความซับซ้อนของการสร้างกราฟตรง ๆ (graph generation) ที่มักประสบปัญหากับความสมเหตุสมผลของพันธะและข้อจำกัดเชิงเคมี อีกทั้งยังรองรับการปรับแต่งเป้าหมายเชิงคุณสมบัติ ทำให้กระบวนการออกแบบโมเลกุลสามารถเปลี่ยนจากการลองผิดลองถูกเป็นการค้นหาเชิงนำทางด้วยโมเดลได้อย่างเป็นระบบ
ข้อพิจารณาด้านการใช้งานเชิงธุรกิจและความท้าทาย
สำหรับสตาร์ทอัพเช่น DeNovoDrug การผสาน GNN และ diffusion models มีศักยภาพลดเวลาค้นพบยาจากปีหลายปีเหลือเป็นเดือน/ไตรมาส โดยเฉพาะในขั้นตอนการออกแบบโมเลกุลนำร่อง อย่างไรก็ตามยังมีความท้าทายที่ต้องบริหารจัดการ เช่น ความน่าเชื่อถือของโมเดลเมื่อขยายสู่เคมีสเปซใหม่ การรับรองว่าผลลัพธ์เคมีสอดคล้องกับการทดลองในห้องแล็บ และต้นทุนคอมพิวต์สำหรับการสาธิตหลายพันตัวอย่าง
สรุป: การรวมกันของ Diffusion Models ที่เชี่ยวชาญด้านการสร้างตัวอย่างต่อเนื่องและการเรียนรู้การ denoising กับ Graph Neural Networks ที่จับโครงสร้างเชิงสัมพันธ์ของโมเลกุลได้อย่างแม่นยำ จึงเป็นกรอบงานที่ทรงพลังสำหรับการออกแบบโมเลกุลต้านไวรัส — ให้ทั้งความสามารถในการสำรวจพื้นที่โมเลกุลอย่างกว้างและการควบคุมคุณสมบัติเชิงเคมีที่จำเป็นต่อการพัฒนายาที่มีประสิทธิภาพ
สถาปัตยกรรมและ pipeline ของ DeNovoDrug
ภาพรวมสถาปัตยกรรมของแพลตฟอร์ม DeNovoDrug
สถาปัตยกรรมของ DeNovoDrug ถูกออกแบบให้เป็นระบบโมดูลาร์ที่ผสาน Diffusion Model สำหรับการสร้างโครงสร้างโมเลกุลกับ Graph‑Neural Networks (GNN) เพื่อการแทนและประเมินสมบัติแบบกราฟ จุดประสงค์หลักคือการย่นระยะเวลาการค้นพบยาจากปีให้เหลือเพียงไม่กี่เดือนด้วยกระบวนการที่เป็นไปโดยอัตโนมัติ ตั้งแต่การนำเข้าข้อมูล (data ingestion) การฝัง (embedding) การฝึกโมเดล (training) ไปจนถึงการคัดกรองเชิงคอมพิวเตอร์ (in‑silico validation) และการส่งมอบตัวอย่างคุณภาพสูงให้กับห้องปฏิบัติการ
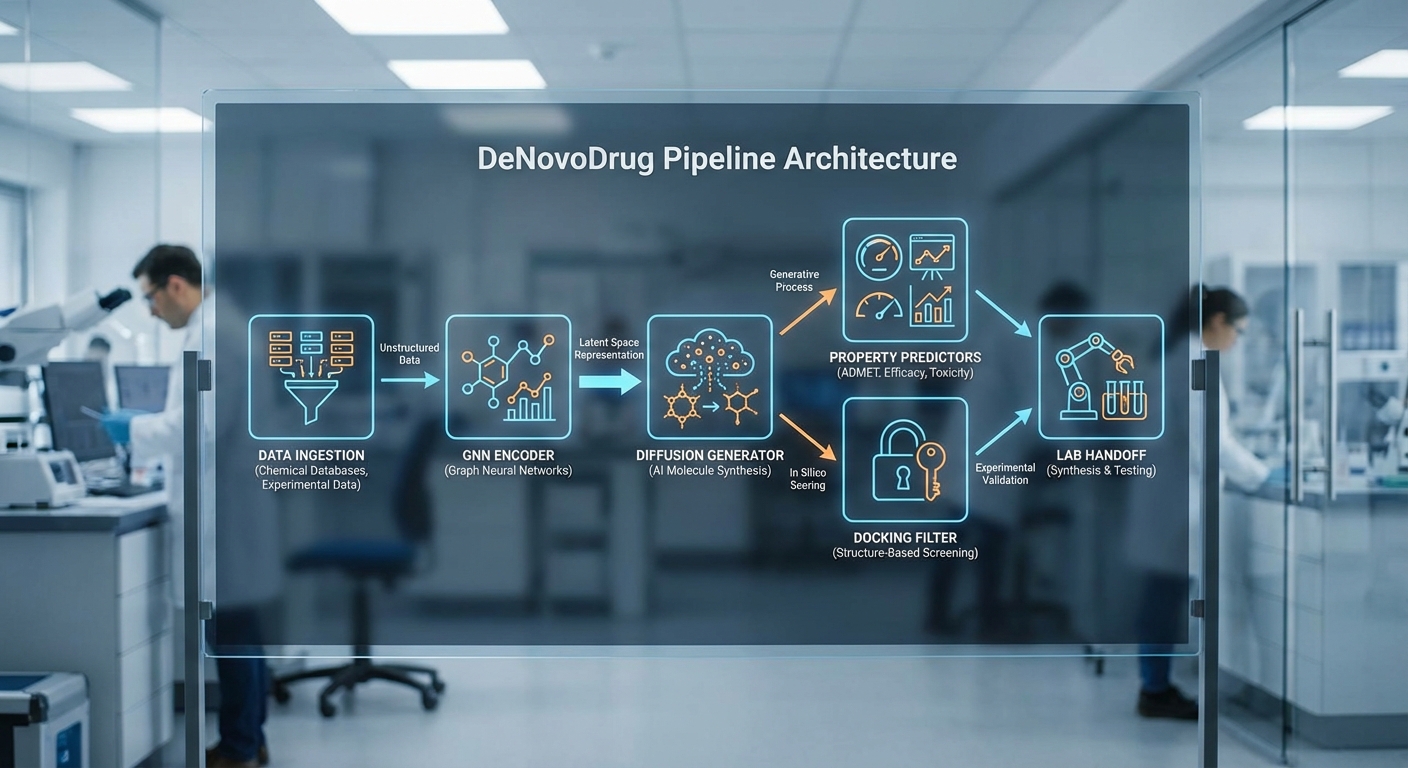
Data ingestion และการแทนโมเลกุล (SMILES / Graph / 3D conformer)
ขั้นตอนแรกเริ่มจากการรวบรวมและทำความสะอาดข้อมูลจากฐานข้อมูลสาธารณะและเอกชนที่คัดกรองแล้ว เช่น ChEMBL, PubChem และชุดข้อมูลการทดสอบต่อไวรัส (viral‑target assays) ของพันธมิตรเชิงอุตสาหกรรม โดยแพลตฟอร์มมีระบบ QC/Audit ที่กรองข้อมูลซ้ำ ข้อมูลที่ขาดหาย และตัวอย่างที่มีค่าผิดปกติ ก่อนจะผ่านการ augmentation เช่น การเพิ่ม conformer 3D ด้วยเครื่องมืออย่าง RDKit/OMEGA, การขยายชุดข้อมูลด้วยการทำ SMILES augmentation (canonical ↔ non‑canonical), และการสังเคราะห์ข้อมูลจำลองจากการผสมโมเลกุล (in silico mutational augmentation) เพื่อเพิ่มความหลากหลายของฝึกสอน
การแทนโมเลกุลรองรับหลายรูปแบบ: SMILES สำหรับสตรีมเวิร์กที่เบา, graph ที่เป็นโหนด‑ขอบ (atom‑bond) สำหรับ GNN และชุด conformer 3D ที่ใช้เสริมข้อมูลเชิงเรขาคณิตกับโมดูลฟีเจอร์เชิงตำแหน่ง (positional features) ระบบสามารถสกัด fingerprint แบบคลาสสิก (เช่น ECFP) พร้อมกับฝังแบบเรียนรู้ได้ (learned embeddings) เพื่อให้โมเดล diffusion ถูกเงื่อนไขด้วยข้อมูลเป้าหมายทางชีวภาพ (target fingerprint) ได้อย่างยืดหยุ่น
Model stack: Encoder (graph embedding) → Diffusion generator → Property predictor → Reranker
สแต็กโมเดลหลักแบ่งออกเป็นหลายชั้น:
- Encoder / Graph embedding: ใช้ GNN ประเภท Message‑Passing (เช่น GIN/MPNN/Graph Transformer) เพื่อแปลงโมเลกุลเป็นเวกเตอร์ embedding ที่รวมข้อมูลธาตุ พันธะ และคุณสมบัติ 3D เช่นระยะเชิงมุมและทรงกลมของ conformer embeddings เหล่านี้ยังสามารถรวม embedding ของโปรตีนเป้าหมายหรือฟีเจอร์ assay เพื่อการเงื่อนไขต่อไป
- Diffusion generator (conditioned): โมเดล diffusion ถูกฝึกให้สร้างกราฟโมเลกุลใหม่โดยถูกเงื่อนไขด้วย target fingerprint/target embedding ซึ่งช่วยกำหนดทิศทางการออกแบบโมเลกุลให้มีแนวโน้มผูกกับเป้าหมายไวรัส โมเดลรองรับทั้งการสร้างจาก latent space และการสร้างกราฟแบบ iterative บนโครงสร้าง atom‑bond โดยใช้ loss เฉพาะทางที่ผสานทั้งความสมจริงของโครงสร้างและความสอดคล้องเชิงสมบัติ
- Property predictor: โมดูลทำนายสมบัติสำคัญแบบพุ่งตรง (เช่น IC50/logP/QED, solubility, permeability, CYP inhibition) โดยใช้ ensemble ของ GNN และ MLP เพื่อให้การทำนายมีความเสถียร ข้อมูลตัวอย่างเช่นระบบมักคัดทิ้งมากกว่า 80% ของโมเลกุลที่มีค่าทำนายไม่เป็นไปตามเกณฑ์ก่อนจะเข้าสู่ขั้นตอน docking
- Reranker (docking + ML‑ADMET): รายชื่อโมเลกุลที่ผ่านการคัดกรองเชิงสมบัติจะถูกส่งเข้าสู่รัน docking (เช่น AutoDock Vina / commercial engines) ร่วมกับตัวประเมิน ADMET เชิงเครื่อง (ML‑based ADMET) และเกณฑ์เช่น Lipinski/Veber/QED เพื่อให้ได้รายการ high‑quality candidates ในระดับที่พร้อมส่งออกห้องปฏิบัติการ
การฝึกสอน (Training) และการประเมินผล
การฝึกสอนรวมทั้งการ pretrain encoder บนชุดข้อมูลขนาดใหญ่ (ตัวอย่าง: หลายล้านโมเลกุลจาก PubChem/ChEMBL) และการ fine‑tune diffusion model กับชุดข้อมูลเฉพาะเป้าหมายไวรัส การฝึกใช้เทคนิคเช่น curriculum learning และ contrastive objectives เพื่อให้ embedding มีความจำเพาะต่อความสามารถในการผูกเป้าหมาย นอกจากนี้ระบบมีการประเมินเชิงปริมาณด้วยเมตริกหลากหลาย เช่น validity/novelty/uniqueness ของโมเลกุล และ predicted potency distribution — เพื่อให้ทีมสามารถติดตามความคืบหน้าและปรับพารามิเตอร์ได้ทันที
Orchestration: จาก generation → in‑silico validation → ส่งมอบให้ห้องปฏิบัติการ
แพลตฟอร์มทำงานบนโครงสร้าง orchestration อัตโนมัติที่ผสาน Kubernetes/Apache Airflow สำหรับการจัดลำดับงาน ตั้งแต่การนำเข้าข้อมูล การฝัง การสร้าง การคัดกรอง และการรัน docking แบบขนาน โดยมี pipeline ที่ลดเวลาขั้นตอนซ้ำซ้อน ตัวอย่างของสเต็ปเชิงลอจิกคือ:
- Data ingestion → featurization (SMILES/graph/3D) → store in feature store
- Batch generation โดย diffusion generator (เช่น 10k–100k candidates ต่อรอบ) → preliminary property prediction
- Filter ตาม thresholds → parallel docking + ML‑ADMET → reranking
- Output รายการ top N (เช่น 50–200) ที่มีคะแนนรวมสูงสุด พร้อมรายงานสรุป (predicted potency, docking score, ADMET flags, synthetic accessibility)
- ส่งมอบข้อมูลเชิงเทคนิคให้ห้องปฏิบัติการในรูปแบบที่พร้อมสำหรับการสังเคราะห์: SMILES, 3D conformers, suggested synthetic routes และ priority tags
ระบบ orchestration ยังรองรับการวนลูปแบบ closed‑loop: ผลการทดลองทางห้องปฏิบัติการ (in vitro/in vivo) ถูกนำกลับมาเป็นข้อมูลฝึกเพิ่มเติม (active learning) ทำให้โมเดลปรับตัวและเพิ่มอัตราความสำเร็จของ candidates ในรอบถัดไป ซึ่งลดเวลาและต้นทุนการค้นพบยาอย่างมีนัยสำคัญ
ตัวอย่างผลลัพธ์และตัวชี้วัดเชิงปฏิบัติ
ในเชิงปฏิบัติ DeNovoDrug รายงานว่า pipeline สามารถผลิตชุด candidate เริ่มต้นจำนวนมากแล้วคัดกรองจนได้รายชื่อคุณภาพสูงภายในไม่กี่สัปดาห์ — โดยตัวอย่างเมตริกที่มักใช้ในการวัดความสำเร็จประกอบด้วย:
- Throughput การสร้าง: ประมาณ 10^4–10^5 โมเลกุลต่อรอบ batch (ปรับตามทรัพยากร)
- อัตราการผ่านขั้นตอน property filter เบื้องต้น: ประมาณ 10–20%
- จำนวน candidates ที่ส่งให้ห้องปฏิบัติการต่อโปรเจ็กต์: ปกติ 50–200 รายการพร้อมข้อมูล ADMET และ synthetic plan
- ระยะเวลาตั้งแต่เริ่มโปรเจ็กต์จนถึงการได้รับชุดตัวอย่างเพื่อทดสอบ: โดยทั่วไปลดจากปีเป็นไม่กี่เดือน โดยมีการวนปรับปรุงผ่าน active learning
สถาปัตยกรรมและ pipeline ดังกล่าวเน้นความสามารถในการปรับขนาด ทำซ้ำได้ และเชื่อมต่อกับกระบวนการทางห้องปฏิบัติการ ซึ่งเป็นหัวใจสำคัญที่ช่วยให้ DeNovoDrug ตอบโจทย์ความต้องการเชิงธุรกิจและเชิงวิทยาศาสตร์ในการพัฒนาโมเลกุลต้านไวรัสอย่างรวดเร็วและมีประสิทธิภาพ
ชุดข้อมูล การฝึก และเมตริกที่ใช้ประเมินผล
ชุดข้อมูล การฝึก และเมตริกที่ใช้ประเมินผล
การออกแบบโมเลกุลต้านไวรัสด้วยโมเดลเชิงสร้างสรรค์อย่างการรวม Diffusion Model กับ Graph‑NN จำเป็นต้องพึ่งพาชุดข้อมูลหลากหลายชนิดทั้งเชิงโครงสร้าง เชิงปฏิกิริยา และเชิงคุณสมบัติทางเภสัชกรรม เพื่อให้โมเดลเรียนรู้ความสัมพันธ์ระหว่างโครงสร้างโมเลกุลและการจับเป้าหมาย ตัวอย่างชุดข้อมูลที่มักถูกใช้ประกอบด้วย:
- Ligand‑target interactions: แหล่งข้อมูลเช่น ChEMBL และ PubChem BioAssay ให้ค่าความสัมพันธ์เช่น IC50, Ki, Kd ซึ่งเป็นสัญญาณหลักสำหรับการทำนายความสามารถในการจับเป้าหมาย
- Bioassay และเฉพาะโรค/ไวรัส: ชุดข้อมูลการทดสอบ in vitro ของไวรัส เช่นชุดข้อมูล SARS‑CoV‑2 จากหน่วยงานสาธารณะและการตีพิมพ์ (เช่น NCATS, COVID‑19 OpenData) ซึ่งให้ผลเชิงชีวภาพตรงต่อการตรวจหา antivirals
- โครงสร้างโปรตีน: ข้อมูลโครงสร้างสามมิติจาก PDB สำหรับการทำ docking และเรียนรู้ลักษณะพ็อกเก็ตของเป้าหมาย
- ข้อมูลคุณสมบัติและ ADMET: ฐานข้อมูลเกี่ยวกับความเป็นพิษ (Ames, hERG), เมตาบอลิซึม, การดูดซึม และคุณสมบัติทางกายภาพ‑เคมี (logP, MW) จากแหล่งสาธารณะและเชิงพาณิชย์
การจัดแบ่งชุดข้อมูลเพื่อนำมาใช้ฝึกและประเมินผลต้องคำนึงถึงการเลียนแบบสถานการณ์จริง เพื่อหลีกเลี่ยงการรั่วไหลของข้อมูลและการประเมินที่เกินจริง แนวปฏิบัติที่ใช้บ่อยได้แก่:
- Train / Validation / Test split: แบ่งแบบมาตรฐาน (เช่น 80/10/10) สำหรับโมเดลทั่วไป
- Scaffold‑split: แยกตามโครงสร้างหลักของโมเลกุล (scaffold) เพื่อทดสอบความสามารถของโมเดลในการทั่วไปไปยังเคมีที่ไม่เคยเห็นมาก่อน
- Time‑split หรือ prospective split: แยกข้อมูลตามลำดับเวลาเพื่อจำลองสถานการณ์การค้นพบยาในอนาคต และทดสอบความทนทานต่อการเปลี่ยนแปลงของข้อมูล
- Cross‑validation และ external benchmarks: ใช้ k‑fold CV, รวมถึงการประเมินซ้ำบนชุดข้อมูลอิสระ (เช่น DUD‑E, LIT‑PCBA) เพื่อยืนยันความเสถียรของโมเดล
- Negative sampling และ data augmentation: สร้างตัวอย่างลบและเปลี่ยนรูปเพื่อปรับสมดุลคลาสและเพิ่มความหลากหลายของเคมี
การประเมินคุณภาพของโมเลกุลและประสิทธิภาพของเวิร์กโฟลว์ประกอบด้วยเมตริกเชิงโมเลกุล เชิงการจับผูก และเชิงปฏิบัติจริง ดังนี้:
- Docking score: ค่าเชิงกลศาสตร์ที่ได้จากการ docking (เช่น AutoDock Vina) โดยทั่วไป threshold นิยมใช้คือ score < −7 kcal/mol เพื่อคัดกรองเบื้องต้น โดยโมเลกุลที่ได้ score ต่ำกว่าจะมีแนวโน้มจับได้ดีกว่า
- Binding affinity prediction (ΔG / pIC50): โมเดลหรือการคำนวณเชิงฟิสิกส์เช่น MM/GBSA ใช้วัดความแข็งแรงการจับ เป้าหมายปกติเช่น IC50 < 1 µM หรือ ΔG < −9 kcal/mol จะถูกมองว่าเป็นน่าสนใจ
- คุณภาพทางเภสัชกรรม: QED (Quantitative Estimate of Drug‑likeness) — ค่า 0–1 โดยค่า >0.4–0.6 มักถูกพิจารณาว่ารับได้, SA score (Synthetic Accessibility) — มาตราส่วน 1–10 โดยค่าน้อยสะท้อนการสังเคราะห์ได้ง่ายกว่า (ตัวอย่าง: SA ≤ 6 เป็นที่ยอมรับ)
- In silico toxicity predictions: แบบจำลองคาดการณ์ผลลัพธ์เช่น Ames mutagenicity, hERG inhibition, hepatotoxicity เพื่อคัดกรองตั้งแต่ระยะคอมพิวเตชัน (ตัวอย่าง: คัดออกหากมีความเสี่ยง hERG สูงหรือ Ames positive)
- Generative model metrics: ความถูกต้องของโครงสร้าง (validity), ความไม่ซ้ำกัน (uniqueness), ความใหม่ (novelty), และการกระจายของคุณสมบัติเทียบกับชุดข้อมูลอ้างอิง เช่น Fréchet ChemNet Distance (FCD)
- การจัดลำดับเชิงปรับแต่งหลายเป้าหมาย: การใช้ Pareto‑front หรือ multi‑objective scoring เพื่อสมดุลระหว่าง affinity, QED, SA และความเป็นพิษ
นอกจากเมตริกเชิงคำนวณแล้ว ทีมพัฒนายังให้ความสำคัญกับตัวชี้วัดทางปฏิบัติซึ่งสะท้อนมูลค่าทางธุรกิจและความเร็วในการพัฒนา:
- จำนวน candidate ที่ส่งตรวจจริง: ปกติแคมเปญจะส่งตัวอย่างตั้งแต่ 50–200 โมเลกุลให้ห้องปฏิบัติการทดสอบ ระดับนี้ช่วยให้คุ้มค่าทั้งในด้านเวลาและงบประมาณ
- Hit rate ในการทดสอบ in vitro: อัตราการพบสารที่มี activity ตามเกณฑ์ (ตัวอย่าง IC50 < 1 µM) — สำหรับการสุ่มจะอยู่ที่ ~0.1–1% ขณะที่การคัดกรองด้วยโมเดลที่ดีอาจเพิ่มเป็น 5–20% หรือสูงกว่า ขึ้นกับความเข้มงวดของเกณฑ์
- Time‑to‑hit (เวลาตั้งแต่เริ่มจนได้ candidate แรก): ความสำเร็จเชิงการตลาดมักวัดจากช่วงเวลา ตัวอย่างจากเทคโนโลยี generative ที่ผสานกับการคัดกรองอัตโนมัติสามารถลดเวลาจากหลายปี (2–3 ปีในกระบวนการแบบดั้งเดิม) เหลือเป็น ไม่กี่เดือน (2–6 เดือน) จนได้ candidate แรกสำหรับทดสอบ
- Enrichment metrics: เช่น Enrichment Factor (EF) ที่ top 1%/5% และ AUROC/PR‑AUC สำหรับโมเดลทำนายกิจกรรม ซึ่งใช้วัดความสามารถในการเรียงลำดับสารที่เป็น active ให้อยู่ด้านบน
- ต้นทุนและ throughput: ค่าใช้จ่ายต่อ candidate และเวลาในการสังเคราะห์/จัดส่งมีผลต่อการตัดสินใจคัดเลือก — การคัดกรองชั้นแรกด้วย computational filters ช่วยลดต้นทุนอย่างมีนัยสำคัญ
การประเมินเชิงปฏิบัติจึงเป็นการผสมผสานหลายชั้น: เริ่มจากเมตริกเชิงคุณภาพโมเลกุล (QED, SA) และการทำนายความเป็นพิษ, ตามด้วยการทดสอบการจับด้วย docking/MM‑GBSA และสุดท้ายคือการยืนยันเชิงห้องทดลอง (in vitro). กลยุทธ์นี้ช่วยให้แพลตฟอร์มอย่าง DeNovoDrug สามารถส่งมอบ ผู้สมัครยาที่มีความเป็นไปได้สูง ให้กับพันธมิตรเชิงพาณิชย์ได้อย่างรวดเร็ว มีความคุ้มค่า และสามารถรายงานตัวชี้วัดเชิงธุรกิจเช่น hit rate และ time‑to‑hit ซึ่งเป็นข้อมูลสำคัญต่อการตัดสินใจลงทุนและการวางแผนเชิงกลยุทธ์
กรณีศึกษา: ออกแบบโมเลกุลต้านไวรัสด้วย DeNovoDrug
กรณีศึกษา: ออกแบบโมเลกุลต้านไวรัสด้วย DeNovoDrug
ในกรณีศึกษาจำลองนี้ สตาร์ทอัพจากสิงคโปร์รายงานกระบวนการใช้แพลตฟอร์ม DeNovoDrug ซึ่งผสาน Diffusion Model กับ Graph‑NN เพื่อออกแบบโมเลกุลต้านไวรัสตั้งแต่การสร้าง (generation) จนถึงรายชื่อผู้สมัครยา (candidates) ที่พร้อมเข้าสู่การสังเคราะห์และทดสอบทางห้องปฏิบัติการ ตัวอย่างผลลัพธ์สำคัญที่รายงานได้แก่ การสร้างโมเลกุลจำนวนมากภายในเวลาจำกัด และการคัดกรองเชิงคุณภาพ-เชิงปริมาณที่นำไปสู่การคัดเลือกสุดท้าย 50 ตัวอย่างที่มีศักยภาพสูง
ขั้นตอนสำคัญและตัวเลขสรุปเชิงปริมาณ (ตัวอย่างจำลอง): ระบบสร้างได้ 100,000 โมเลกุลภายใน 48 ชั่วโมง จากนั้นใช้ตัวกรองเชิงสภาพและเชิงคุณภาพ เช่น QED (Quantitative Estimate of Drug-likeness) และ SA (Synthetic Accessibility) ลดจำนวนนั้นเหลือ 1,200 โมเลกุล เพื่อเข้าสู่การคัดกรองด้วย docking และการประเมินค่าความเป็นไปได้เชิงชีวภาพด้วย Graph‑NN ผลการ docking เบื้องต้นคัดเลือกได้ 50 candidates ที่มีคะแนนดีพอสมควรสำหรับขั้นตอนถัดไป
ระยะเวลาโดยรวมตั้งแต่การ generation จนได้รายชื่อ 50 candidates ใช้เวลาเฉลี่ย 2–8 สัปดาห์ ขึ้นกับความเข้มข้นของการคัดกรองและทรัพยากรคำนวณ ในขณะที่กระบวนการแบบดั้งเดิม (รวมการออกแบบแบบแมนนวล การสังเคราะห์เชิงทดลอง และการคัดกรองระดับแรก) อาจใช้เวลาเป็นปีหรือมากกว่า ทำให้ DeNovoDrug ช่วยลดเวลาค้นพบยาจากปีเหลือเพียงไม่กี่สัปดาห์ถึงสองเดือนในขั้นตอนคิดค้นเบื้องต้น
ตัวชี้วัดเชิงตัวเลขที่รายงาน (ตัวอย่างเชิงสถิติจากชุด 1,200 โมเลกุลก่อนคัดเลือกสุดท้าย):
- Distribution ของ docking scores (AutoDock Vina แบบจำลอง): ช่วงคะแนนจาก -10.8 ถึง -6.2 kcal/mol ค่าเฉลี่ย ≈ -8.4 kcal/mol ค่ามัธยฐาน ≈ -8.2 kcal/mol
- Predicted pIC50 (จาก Graph‑NN): ช่วงประมาณ 4.2 – 7.2 ค่าเฉลี่ย ≈ 6.1 มัธยฐาน ≈ 6.4 (ค่าสูงสุดตัวอย่างหนึ่งที่คาดการณ์ได้คือ pIC50 ≈ 7.2 ซึ่งสอดคล้องกับ IC50 ประมาณ 63 nM)
- ความหลากหลายโครงสร้าง: ค่าเฉลี่ย pairwise Tanimoto similarity ≈ 0.32 (แสดงความหลากหลายสูง) จำนวน Bemis‑Murcko scaffolds ที่ไม่ซ้ำกันในชุด 1,200 ≈ 420 (top 10 scaffolds ครอบคลุม ~18% ของชุด)
ผลลัพธ์เชิงปฏิบัติการเบื้องต้นหลังการคัดเลือก 50 candidates (ตัวอย่างการดำเนินการต่อ):
- จาก 50 candidates ทีมคัดเลือกเชิงกลยุทธ์ได้ 12 โมเลกุล เพื่อสังเคราะห์ตามลำดับความสำคัญ
- สังเคราะห์สำเร็จและทดสอบในห้องปฏิบัติการเบื้องต้นจำนวน 8 โมเลกุล
- ผลการทดสอบ in vitro แรกพบว่า 3 โมเลกุล มี activity เด่น (pIC50 ≥ 6; IC50 ≤ 1 µM) และมี 1 โมเลกุล เป็น lead ที่มี pIC50 ≈ 7.2 (IC50 ~63 nM)
การสรุปเชิงธุรกิจ: กรณีศึกษานี้แสดงให้เห็นความสามารถของแพลตฟอร์ม DeNovoDrug ในการเร่งวงจรการค้นพบยาตั้งแต่การสร้างโมเลกุลจำนวนมาก การกรองเชิงคุณภาพและเชิงคำนวณ จนถึงการได้รายชื่อ candidates ที่มีโอกาสประสบความสำเร็จในเชิงทดลองจริง ภายใต้ทรัพยากรและระยะเวลาที่จำกัด ผลลัพธ์เชิงตัวเลขข้างต้นเป็นตัวอย่างจำลองที่สื่อถึงศักยภาพในการลดเวลาและต้นทุนของกระบวนการค้นพบยาโดยรวม
การประเมินความปลอดภัย กฎระเบียบ และข้อจำกัด
การประเมินความปลอดภัย กฎระเบียบ และข้อจำกัด
แม้แพลตฟอร์มอย่าง DeNovoDrug ที่ผสาน diffusion model กับ Graph‑NN จะสามารถเร่งกระบวนการค้นหาผู้สมัครยาได้อย่างมีนัยสำคัญ — ย่นระยะเวลาจากปีเป็นเดือนสำหรับการค้นหาเบื้องต้น — แต่ ขั้นตอนทางวิทยาศาสตร์และกฎระเบียบยังคงเป็นอุปสรรคสำคัญก่อนการนำไปใช้จริง งานวิจัยและรายงานหลายฉบับชี้ให้เห็นว่าขั้นตอนการค้นพบยาแบบดั้งเดิมใช้เวลาหลายปี (โดยทั่วไป 10–15 ปี) และมีต้นทุนรวมตั้งแต่หลายร้อยล้านไปจนถึงหลายพันล้านดอลลาร์สหรัฐสำหรับการพัฒนาจนถึงการขึ้นทะเบียน การใช้ AI เพียงอย่างเดียวไม่อาจแทนการทดสอบในห้องปฏิบัติการ (in vitro) และการทดสอบในสิ่งมีชีวิต (in vivo) ที่จำเป็นสำหรับการประเมินความปลอดภัย ประสิทธิภาพ และความสามารถในการผลิตเชิงอุตสาหกรรมได้
การตรวจสอบความปลอดภัยทางชีวภาพและขั้นตอนการทดสอบ เป็นข้อกำหนดที่หน่วยงานกำกับดูแล เช่น FDA (สหรัฐฯ), EMA (ยุโรป) และ HSA (สิงคโปร์) ยืนยันว่ายาใหม่ต้องผ่านการทดสอบ GLP ในห้องปฏิบัติการ การศึกษาพิษศาสตร์ก่อนคลินิก การประเมิน ADME (absorption, distribution, metabolism, excretion) และขั้นตอนการตรวจสอบคุณภาพผลิตภัณฑ์เชิงอุตสาหกรรม (GMP) ก่อนการยื่น IND/CTA เพื่อเริ่มการทดลองทางคลินิก ตัวอย่างการประเมิน เช่น การทดสอบ hepatotoxicity, cardiotoxicity, และการสะสมในเนื้อเยื่อต่าง ๆ มักเป็นสาเหตุหลักที่ทำให้ผู้สมัครยาล้มเหลวในระยะคลินิก — งานวิเคราะห์หลายชิ้นระบุว่า ประมาณร้อยละ 30–40 ของการล้มเหลวในระยะคลินิกสัมพันธ์กับปัญหาความเป็นพิษหรือ ADMET ซึ่งย้ำว่าโมเดลคอมพิวเตอร์ต้องถูกยืนยันด้วยข้อมูลเชิงประจักษ์เสมอ
ความเสี่ยงจาก bias ในข้อมูลและความเสี่ยงเชิงชีวภาพ เป็นอีกประเด็นสำคัญที่ต้องพิจารณาอย่างรอบคอบ หากชุดข้อมูลฝึก (training datasets) มีความเบ้ (biased) เช่น เนื้อหาของสารเคมีที่มาจากกลุ่มโครงสร้างเพียงไม่กี่ชนิด หรือการขาดข้อมูลจากแหล่งที่มีความหลากหลายทางชีวภาพ โมเดลอาจสร้างโมเลกุลที่ดูดีบนกระดาษแต่มีคุณสมบัติไม่พึงประสงค์ เช่น ความเป็นพิษสูง, เมตาบอไลซ์สร้างสารพิษ, หรือมีการสังเคราะห์ที่ยากในทางปฏิบัติ นอกจากนี้ยังมีความเสี่ยงด้านความปลอดภัยของข้อมูลเชิงชีวภาพ (biosecurity) — ข้อมูลลำดับ ชุดข้อมูล assay หรือโปรโตคอลการทดลองที่หลุดออกไปอาจถูกนำไปใช้ในทางที่ก่อให้เกิดอันตรายหรือการละเมิดจริยธรรมได้ การกำกับดูแลด้านการเข้าถึงข้อมูล การจัดการสิทธิความเป็นเจ้าของ และการประเมินความเสี่ยงแบบ dual‑use จึงเป็นสิ่งจำเป็น
ข้อจำกัดเชิงเทคนิคของโมเดล ต้องยอมรับว่าเทคโนโลยีปัจจุบันยังมีข้อด้อยหลายประการ:
- ความแม่นยำของการจำลองการจับกัน (binding) — วิธี docking แบบรวดเร็วมักให้ผลคร่าว ๆ เท่านั้น ขณะที่วิธีที่มีความแม่นยำสูงกว่า เช่น free energy perturbation (FEP) หรือ molecular dynamics (MD) ต้องใช้ความสามารถคำนวณระดับสูงและเวลาในการคำนวณมาก การพยากรณ์พลังงานการจับกันอย่างแม่นยำในสภาวะแวดล้อมชีวภาพจริงจึงยังเป็นความท้าทาย
- การทำนาย ADMET อย่างเชื่อถือได้ — แบบจำลอง ML/AI มักถูกจำกัดโดยขนาดและความหลากหลายของข้อมูลยืนยันทางคลินิก การทำนายผลข้างเคียงหายาก (rare adverse events) หรือปฏิกิริยาทางเภสัชจลนศาสตร์เฉพาะกลุ่มประชากรยังมีความไม่แน่นอนสูง
- ข้อจำกัดด้าน compute และต้นทุน — การฝึกโมเดลระดับใหญ่และการรันวิเคราะห์ความแม่นยำสูงต้องอาศัย GPU/TPU หรือทรัพยากร HPC ที่มีต้นทุนพลังงานและการเงินสูง ซึ่งอาจจำกัดการทดสอบเชิงลึกและการทำซ้ำผลในสเกลอุตสาหกรรม
- ความสามารถในการสังเคราะห์จริง (synthetic accessibility) — โมเดลสร้างโมเลกุลได้มากมาย แต่โมเลกุลที่ได้อาจยากหรือแพงต่อการสังเคราะห์จริง ซึ่งจำเป็นต้องประเมินร่วมกับเครื่องมือวางแผนการสังเคราะห์ (retrosynthesis planners) และความเชี่ยวชาญทางเคมี
เพื่อบรรเทาความเสี่ยงเหล่านี้ ควรนำแนวปฏิบัติดังต่อไปนี้มาใช้ควบคู่กับการพัฒนา AI: การคัดกรองและแยกประเภทข้อมูลอย่างเข้มงวด การประเมินความไม่แน่นอน (uncertainty quantification) และการทดสอบแบบ retrospective/independent benchmark รวมถึงการมีมนุษย์ตรวจสอบผล (human‑in‑the‑loop) การทำงานร่วมกับห้องปฏิบัติการ GLP และการปรึกษาหารือกับหน่วยงานกำกับดูแลตั้งแต่ระยะเริ่มต้น ตัวอย่างเช่น บริษัทบางแห่งที่ใช้ AI เช่น Exscientia เคยรายงานผู้สมัครยาที่เกิดจากกระบวนการที่มี AI เข้าร่วมเข้าสู่การทดลองทางคลินิก ซึ่งเป็นสัญญาณบ่งชี้ว่าการผสมผสานนี้มีศักยภาพจริง แต่ก็ยังต้องผ่านกระบวนการยืนยันเชิงทดลองและกฎระเบียบอย่างเคร่งครัดก่อนการใช้งานในวงกว้าง
สรุปคือ แม้ DeNovoDrug จะเป็นเครื่องมือเปลี่ยนเกมสำหรับการค้นพบโมเลกุลต้านไวรัส แต่การนำผลลัพธ์ไปสู่การใช้งานจริงจำเป็นต้องผ่านการประเมินความปลอดภัย การทดสอบทางคลินิก และการควบคุมทางกฎระเบียบอย่างครบถ้วน พร้อมทั้งการจัดการกับความเสี่ยงด้านข้อมูล ความไม่แน่นอนของโมเดล และข้อจำกัดด้านเทคนิคอย่างเป็นระบบ เพื่อให้ผลลัพธ์ที่ได้มีความน่าเชื่อถือ ปลอดภัย และสอดคล้องกับมาตรฐานสาธารณสุขสากล
คู่มือเชิงปฏิบัติสำหรับนักพัฒนาและนักวิจัย
คู่มือเชิงปฏิบัติสำหรับนักพัฒนาและนักวิจัย
บทต่อไปนี้มุ่งให้แนวทางเชิงปฏิบัติสำหรับการนำแนวคิดของ DeNovoDrug — การผสาน Diffusion Model กับ Graph Neural Network (GNN) — มาทดลองและพัฒนาเอง ตั้งแต่การตั้งค่าสแต็กซอฟต์แวร์ ข้อมูลตัวอย่าง ทรัพยากรคอมพิวต์ เทคนิคการฝึก ไปจนถึง pipeline สำหรับการคัดกรองและตรวจสอบผลเชิง in silico เพื่อเร่งวงจรการค้นพบยาอย่างมีประสิทธิภาพ เหมาะสำหรับทีมนักพัฒนา นักวิจัย และผู้บริหารฝ่าย R&D ที่สนใจนำไปปรับใช้เชิงพาณิชย์
สแต็กซอฟต์แวร์ที่แนะนำ
- Frameworks: PyTorch (แนะนำสำหรับชุมชน GNN และความยืดหยุ่น) หรือ JAX (สำหรับการคำนวณประสิทธิภาพสูงบน TPU)
- GNN Libraries: PyTorch Geometric (PyG) หรือ DGL — ทั้งสองรองรับการสร้างกราฟโมเลกุล (node: atoms, edge: bonds) และ mini-batching
- Diffusion / Score-based libraries: Hugging Face Diffusers (ปรับเพื่อโมเลกุล), guided-diffusion, หรือ score_sde_pytorch (Song et al.) — สามารถนำมาขยายให้รองรับกราฟ/latent space
- Chemoinformatics: RDKit สำหรับการแปลง SMILES ↔ graph, คำนวณคุณสมบัติ (logP, QED, SAscore) และสร้างการจับคู่ scaffold
- Docking & in silico tools: AutoDock Vina, OpenMM/GROMACS สำหรับ MD, DeepPurpose หรือ ADMET-predictors สำหรับการพยากรณ์ ADMET
- Experiment tracking & MLOps: Weights & Biases หรือ MLflow สำหรับ logging, model versioning และ hyperparameter sweeps
การตั้งค่าเบื้องต้นและข้อมูลตัวอย่าง
ชุดข้อมูลเริ่มต้น: แนะนำเริ่มจากขนาดเล็ก 10k–100k โมเลกุล เพื่อ iterate ได้เร็ว ตัวอย่างแหล่งข้อมูล: ZINC, ChEMBL, PubChem subsets, BindingDB หรือชุดข้อมูลเฉพาะเช่น MoleculeNet datasets (e.g., ESOL, FreeSolv) หากเน้นไวรัส ให้รวม ligand ของ target viral proteins จาก PDB และ COVID-19 Open datasets
การแบ่งข้อมูล: ใช้ split แบบ 80/10/10 เป็น baseline แต่แนะนำใช้ scaffold split เพื่อทดสอบการทั่วไปของโมเดลทางเคมีจริงจัง (scaffold-based holdout มักทำให้ผลทดสอบสมจริงกว่า random split)
Data preprocessing: ปรับ SMILES ให้ canonical, สร้าง adjacency + feature matrices (atom types, formal charge, hybridization), และ normalized target properties (e.g., pIC50, logP)
โครงร่างโค้ด (high-level) และ workflow
- Step 1: สร้าง GNN encoder/decoder
- ฝึก GNN เพื่อเรียนรู้ embedding ของโมเลกุล (node/graph level) — ตัวอย่างสถาปัตยกรรม: GIN/GraphSAGE + global pooling
- Step 2: สร้าง diffusion process บน latent space
- ใช้ encoder → latent z, ฝึก diffusion model ให้เรียนรู้ p(z_t | z_{t+1}) หรือใช้ score matching บนกราฟ representation
- Step 3: การสร้างแบบมีเงื่อนไข (conditional generation)
- ใช้ classifier guidance หรือ concatenated conditional vectors (เช่น desired pIC50, target fingerprint) ในทุก timestep ของ diffusion
- Step 4: sampling → decode → rerank
- แปลง latent → graph → SMILES, คำนวณคุณสมบัติเบื้องต้น และ rerank ด้วย docking/ADMET ในลูปอัตโนมัติ
ตัวอย่าง pseudo-code (very high-level):
- dataset = load_molecules("ChEMBL_subset")
- gnn = GNNEncoder(in_feats, hid)
- latent = gnn.encode(graph)
- diffusion = ScoreModel(latent_dim)
- for epoch in epochs:
- z0 = latent
- z_t = forward_noise(z0)
- loss = score_matching_loss(diffusion, z_t, cond)
- opt.step()
- samples = sampling_with_classifier_guidance(diffusion, cond)
คำแนะนำด้าน compute และ hyperparameters เบื้องต้น
- GPU/TPU: สำหรับการทดลองเบื้องต้น GPU แบบ single node เช่น NVIDIA RTX 3090 / A5000 / 4090 (24–48 GB) เหมาะ หากต้องการ scale ขึ้น ใช้ A100 40/80 GB หรือ multi-GPU (8×A100) สำหรับ training dataset ขนาดใหญ่และ hyperparameter sweeps ทางเลือกในการใช้ TPU v3/v4 เหมาะกับ JAX
- Memory & batch size: batch size 32–256 ขึ้นกับขนาดโมเดลและ GPU memory; หากจำกัดหน่วยความจำใช้ gradient accumulation
- Diffusion hyperparameters (เริ่มต้น):
- timesteps: 1000 (สามารถลดเป็น 100–250 สำหรับ accelerated samplers เช่น DDIM)
- learning rate: 1e-4 – 5e-5 (AdamW), weight decay 1e-2
- EMA (model average): decay = 0.999
- loss weighting: MSE on score or weighted denoising loss ตาม noise schedule
- GNN hyperparameters: layers 3–6, hidden 128–512, dropout 0.1–0.3
- การฝึก: epochs 50–200 (ขึ้นกับ dataset), early stopping บน validation scaffold-split
เทคนิคการฝึกเชิงปฏิบัติ
- Transfer learning: เริ่มจาก GNN pretrained (เช่น MolCLR, ChemBERTa-style embeddings) เพื่อลดเวลา converging — fine-tune บน task-specific targets (e.g., viral protease ligands)
- Conditional generation: ใช้ classifier guidance (training classifier on property of interest) หรือ concatenated conditioning vectors ใน latent to steer generation towardคุณสมบัติเป้าหมาย เช่น pIC50 > threshold, low SAscore
- Curriculum learning: เริ่มจาก generation ของโมเลกุลง่าย ๆ แล้วค่อยเพิ่มความซับซ้อนของ constraints
- Automated pipeline สำหรับ reranking & in silico validation: ตั้ง workflow ดังนี้ —
- generate → canonicalize (RDKit) → filter (rule-of-5, PAINS) → compute properties (QED, logP, SAscore) → docking (AutoDock Vina) → ADMET predictions → final rerank
- นำผลกลับมาทำ active learning loop: ผลการ docking/MD ใช้เป็น signal สำหรับ fine-tune model (reinforcement/score reweighting)
การทดลองและการติดตามผล
Experiment tracking: ใช้ Weights & Biases หรือ MLflow สำหรับ logging ของ metrics (validation loss, property distributions, docking scores), artifact storage (model checkpoints), และ reproducible runs พร้อม config files (YAML/JSON)
Evaluation metrics: validity (SMILES valid %), uniqueness, novelty (vs train set), property distribution shift, docking score distribution, predicted ADMET flags โดยเฉพาะ scaffold diversity เพื่อหลีกเลี่ยง mode collapse
คำแนะนำเชิงปฏิบัติและข้อควรระวัง
- คุณภาพข้อมูล: ตรวจสอบ duplicates, salts, tautomer canonicalization และทำการ normalization ก่อน training
- ไลเซนส์: ตรวจสอบข้อจำกัดเชิงสิทธิ์ของชุดข้อมูล (เช่น ChEMBL/DrugBank) ก่อนใช้ในโปรเจกต์เชิงพาณิชย์
- ความปลอดภัยและจริยธรรม: สังเกตความเสี่ยงด้านการใช้เพื่อการประดิษฐ์สารอันตราย และปฏิบัติตามแนวทางการวิจัยรับผิดชอบ
แหล่งข้อมูลและเอกสารอ้างอิงที่ควรอ่านเพิ่มเติม
- Song, J., et al., "Score-Based Generative Modeling through Stochastic Differential Equations."
- Ho, J., et al., "Denoising Diffusion Probabilistic Models (DDPMs)"
- Huang, K., et al., "MolCLR: Molecular Contrastive Learning"
- RDKit documentation — https://www.rdkit.org/
- Hugging Face Diffusers — https://huggingface.co/docs/diffusers/
- PyTorch Geometric / DGL documentation
- MoleculeNet benchmark papers and datasets
สรุป: เริ่มจากสแต็กมาตรฐาน (PyTorch + PyG + RDKit + Diffusion library) พร้อม dataset ขนาด 10k–100k สำหรับการ iterate รวดเร็ว ใช้ transfer learning และ conditional guidance เพื่อนำโมเดลไปสู่ผลลัพธ์ที่มีความเป็นไปได้เชิงเภสัชกรรม ตั้ง pipeline อัตโนมัติสำหรับ reranking และ in silico validation เพื่อให้กระบวนการค้นพบยาสามารถย่นเวลาจากปีเหลือเป็นเดือนหรือสัปดาห์ได้อย่างเป็นระบบและปลอดภัย
บทสรุป
แพลตฟอร์ม DeNovoDrug ของสตาร์ทอัพสิงคโปร์แสดงให้เห็นว่า การผสาน diffusion models เข้ากับ Graph‑NN ในกระบวนการออกแบบโมเลกุลสามารถเร่งการค้นพบยาขั้นต้นได้อย่างมีนัยสำคัญ — จากที่ปกติอาจกินเวลาเป็นปีหรือหลายปี สามารถลดลงเป็นระดับเดือนในหลายกรณี ตัวอย่างเช่น ระบบเชิงคำนวณสามารถสร้างและคัดกรอง candidate โมเลกุลต้านไวรัสได้ในรอบเวลาที่สั้นลง และรายงานจากงานวิจัยและโครงการพัฒนาเชิงอุตสาหกรรมชี้ว่าความเร็วในการผลิตแบบจำลองและการค้นพบเบื้องต้นอาจเพิ่มขึ้นเป็นหลายสิบถึงหลายร้อยเปอร์เซ็นต์ อย่างไรก็ตามการประหยัดเวลาและต้นทุนเหล่านี้ยังต้องถูกทดสอบด้วย การยืนยันผลทางห้องปฏิบัติการ (wet‑lab validation) และการปฏิบัติตามกฎระเบียบด้านความปลอดภัยทางชีวภาพและการทดลองทางคลินิกก่อนที่จะนำไปสู่การใช้จริง
นักวิจัยและผู้พัฒนาแพลตฟอร์มควรให้ความสำคัญกับคุณภาพของชุดข้อมูล การประเมินความปลอดภัย (toxicology และ off‑target effects) และการวัดประสิทธิภาพด้วยมาตรฐานที่เข้มงวด เพื่อหลีกเลี่ยงผลลัพธ์ที่ลวงหรือไม่ปลอดภัย การร่วมมือกับภาคเภสัชกรรมและห้องปฏิบัติการเชิงทดลองเป็นกุญแจสำคัญในการเร่งนำ candidate สู่การทดสอบจริง (hit‑to‑lead และ lead optimization) รวมถึงการบูรณาการกับการทดสอบอัตโนมัติและการวิเคราะห์ทางชีวสารสนเทศเพื่อให้วงจรการพัฒนาครบถ้วนและเชื่อถือได้
มุมมองอนาคตชี้ให้เห็นว่าแพลตฟอร์มเช่น DeNovoDrug จะมีบทบาทสำคัญในการเปลี่ยนโครงสร้างการค้นพบยา โดยเฉพาะในภาวะระบาดหรือโรคเกิดใหม่ การผสมผสาน AI ขั้นสูง การทดลองแบบ high‑throughput และความร่วมมือข้ามสถาบันจะช่วยให้การถ่ายทอดจากโมเดลคอมพิวเตอร์สู่การทดสอบทางคลินิกเป็นไปอย่างรวดเร็วและปลอดภัย แต่ความท้าทายด้านความโปร่งใสของโมเดล การตรวจสอบย้อนกลับ (explainability) และกรอบกำกับดูแลที่เหมาะสมยังคงต้องได้รับการพัฒนาอย่างต่อเนื่องเพื่อให้เทคโนโลยีนี้สามารถนำมาใช้ได้อย่างรับผิดชอบและมีประสิทธิผลในวงกว้าง



